Warning
This document is for an in-development version of Galaxy. You can alternatively view this page in the latest release if it exists or view the top of the latest release's documentation.
Data Types¶
Adding a New Data Type (Subclassed)¶
This specification describes the Galaxy source code changes required to add support for a new data type.
Every Galaxy dataset is associated with a datatype
which can be determined by the file extension (or
format in the history item). Within Galaxy, supported
datatypes are contained in the
galaxy.datatypes.registry:Registry class, which has
the responsibility of mapping extensions to datatype
instances. At start up this registry is initialized
with data type values from the datatypes_conf.xml
file. All data type classes are a subclass of the
galaxy.datatypes.data:Data class.
We’ll pretend to add a new datatype format named
Foobar whose associated file extension is foo to
our local Galaxy instance as a way to provide the
details for adding support for new data types. Our
example Foobar data type will be a subclass of
galaxy.datatypes.tabular.Tabular.
Step 1: Register Data Type¶
We’ll add the new data type to the <registration>
tag section of the datatypes_conf.xml file. Sample
<datatype> tag attributes in this section are:
<datatype extension="ab1" type="galaxy.datatypes.images:Ab1" mimetype="application/octet-stream" display_in_upload="true"/>
where
extension- the data type’s Dataset file extension (e.g.,ab1,bed,gff,qual, etc.)type- the path to the class for that data type.mimetype- if present (it’s optional), the data type’s mime typedisplay_in_upload- if present (it’s optional and defaults to False), the associated file extension will be displayed in the “File Format” select list in the “Upload File from your computer” tool in the “Get Data” tool section of the tool panel.
Note: If you do not wish to add extended
functionality to a new datatype, but simply want
to restrict the output of a set of tools to be used
in another set of tools, you can add the flag
subclass="True" to the datatype definition line.
Example:
<datatype extension="my_tabular_subclass" type="galaxy.datatypes.tabular:Tabular" subclass="True"/>
Step 2: Sniffer¶
Galaxy tools are configured to automatically set the
data type of an output dataset. However, in some
scenarios, Galaxy will attempt to determine the data
type of a file using a sniffer (e.g., uploading a
file from a local disk with ‘Auto-detect’ selected in
the File Format select list). The order in which
Galaxy attempts to determine data types is critical
because some formats are much more loosely defined
than others. The order in which the sniffer for each
data type is applied to the file should be most
rigidly defined formats first followed by less and
less rigidly defined formats, with the most loosely
defined format last, and then a default format
associated with the file if none of the data type
sniffers were successful. The order in which data
type sniffers are applied to files is implicit in the
<sniffers> tag set section of the
datatypes_conf.xml file. We’ll assume that the
format of our Foobar data type is fairly rigidly
defined, so it can be placed closer to the start of
the sniff order.
<sniffers>
<sniffer type="galaxy.datatypes.sequence:Maf"/>
<sniffer type="galaxy.datatypes.sequence:Lav"/>
<sniffer type="galaxy.datatypes.tabular:Foobar"/>
Step 3: Data Type Class¶
We’ll now add the Foobar class to
lib/galaxy/datatypes/tabular.py. Keep in mind that
your new data type class should be placed in a file
that is appropriate (based on its superclass), and
that the file will need to be imported by
lib/galaxy/datatypes/registry.py. You will need to
include a file_ext attribute to your class and
create any necessary functions to override the
functions in your new data type’s superclass (in our
example, the galaxy.datatypes.tabular.Tabular class).
In our example below, we have set our class’s
file_ext attribute to “foo” and we have overridden
the __init__(), init_meta() and sniff()
functions. It is important to override functions
(especially the meta data and sniff functions) if the
attributes of your new class differ from those of its
superclass. Note: sniff functions are not required to
be included in new data type classes, but if the sniff
function is missing, Galaxy will call the superclass
method.
from galaxy.datatypes.sniff import get_headers
from galaxy.datatypes.tabular import Tabular
class Foobar(Tabular):
"""Tab delimited data in foo format"""
file_ext = "foo"
MetadataElement(name="columns", default=3, desc="Number of columns", readonly=True)
def __init__(self, **kwd):
"""Initialize foobar datatype"""
Tabular.__init__(self, **kwd)
self.do_something_else()
def init_meta(self, dataset, copy_from=None):
Tabular.init_meta(self, dataset, copy_from=copy_from)
if elems_len == 8:
try:
map(int, [hdr[6], hdr[7]])
proceed = True
except:
pass
def sniff(self, filename):
headers = get_headers(filename, '\t')
try:
if len(headers) < 2:
return False
for hdr in headers:
if len(hdr) > 1 and hdr[0] and not hdr[0].startswith('#'):
if len(hdr) != 8:
return False
try:
map(int, [hdr[6], hdr[7]])
except:
return False
# Do other necessary checking here...
except:
return False
# If we haven't yet returned False, then...
return True
...
That should be it! If all of your code is functionally correct you should now have support for your new data type within your Galaxy instance.
Adding a New Data Type (completely new)¶
Basic Datatypes¶
In this real life example,
we’ll add a datatype named GenBank, to support
GenBank files.
First, we’ll set up a file named csequence.py in
lib/galaxy/datatypes/csequence.py. This file could
contain some of the standard sequence types, though
we’ll only implement GenBank.
"""
Classes for all common sequence formats
"""
from galaxy.datatypes import data
from galaxy.datatypes.metadata import MetadataElement
import os
import logging
log = logging.getLogger(__name__)
class GenBank(data.Text):
"""
abstract class for most of the molecule files
"""
file_ext = "genbank"
This is all you need to get started with a datatype.
Now, load it into your datatypes_conf.xml by adding
the following line:
<datatype extension="genbank" type="galaxy.datatypes.csequence:GenBank" display_in_upload="True" />
and start up your server, the datatype will be available.
Adding a Sniffer¶
Datatypes can be “sniffed”, their formats can be
automatically detected from their contents. For
GenBank files that’s extremely easy to do, the first
5 characters will be LOCUS, according to section
3.4.4 of the specification.
To implement this in our tool we first have to add
the relevant sniffing code to our GenBank class in
csequence.py.
def sniff(self, filename):
header = open(filename).read(5)
return header == 'LOCUS'
and then we have to register the sniffer in
datatypes_conf.xml.
<sniffer type="galaxy.datatypes.csequence:GenBank"/>
Once that’s done, restart your server and try
uploading a genbank file. You’ll notice that the
filetype is automatically detected as genbank once
the upload is done.
More Features¶
One of the useful things your datatype can do is provide metadata. This is done by adding metadata entries inside your class like this:
class GenBank(data.Text):
file_ext = "genbank"
MetadataElement(name="number_of_sequences", default=0, desc="Number of sequences", readonly=True, visible=True, optional=True, no_value=0)
Here we have a MetadataElement, accessible in
methods with a dataset parameter from
dataset.metadata.number_of_sequences. There are a
couple relevant functions you’ll want to override
here:
set_peek(self, dataset, is_multi_byte=False)
set_meta(self, dataset, **kwd)
The set_peek function is used to determine the blurb
of text that will appear to users above the preview
(first 5 lines of the file, the file peek), informing
them about metadata of a sequence. For genbank files,
we’re probably interested in how many genome/records
are contained within a file. To do that, we need to
count the number of times the word LOCUS appears as
the first five characters of a line. We’ll write a
function named _count_genbank_sequences.
def _count_genbank_sequences(self, filename):
count = 0
with open(filename) as gbk:
for line in gbk:
if line[0:5] == 'LOCUS':
count += 1
return count
Which we’ll call in our set_meta function, since
we’re setting metadata about the file.
def set_meta(self, dataset, **kwd):
dataset.metadata.number_of_sequences = self._count_genbank_sequences(dataset.file_name)
Now we’ll need to make use of this in our set_peek
override:
def set_peek(self, dataset, is_multi_byte=False):
if not dataset.dataset.purged:
# Add our blurb
if (dataset.metadata.number_of_sequences == 1):
dataset.blurb = "1 sequence"
else:
dataset.blurb = "%s sequences" % dataset.metadata.number_of_sequences
# Get standard text peek from dataset
dataset.peek = data.get_file_peek(dataset.file_name, is_multi_byte=is_multi_byte)
else:
dataset.peek = 'file does not exist'
dataset.blurb = 'file purged from disk'
This function will be called during metadata setting.
Try uploading a multi record genbank file and testing
it out. If you don’t have a multi-record genbank file,
simply concatenate a single file together a couple
times and upload that.
By now you should have a complete GenBank parser in
csequence.py that looks about like the following:
from galaxy.datatypes import data
from galaxy.datatypes.metadata import MetadataElement
import logging
log = logging.getLogger(__name__)
class GenBank(data.Text):
file_ext = "genbank"
MetadataElement(name="number_of_sequences", default=0, desc="Number of sequences", readonly=True, visible=True, optional=True, no_value=0)
def set_peek(self, dataset, is_multi_byte=False):
if not dataset.dataset.purged:
# Add our blurb
if (dataset.metadata.number_of_sequences == 1):
dataset.blurb = "1 sequence"
else:
dataset.blurb = "%s sequences" % dataset.metadata.number_of_sequences
# Get
dataset.peek = data.get_file_peek(dataset.file_name, is_multi_byte=is_multi_byte)
else:
dataset.peek = 'file does not exist'
dataset.blurb = 'file purged from disk'
def get_mime(self):
return 'text/plain'
def sniff(self, filename):
header = open(filename).read(5)
return header == 'LOCUS'
def set_meta(self, dataset, **kwd):
"""
Set the number of sequences in dataset.
"""
dataset.metadata.number_of_sequences = self._count_genbank_sequences(dataset.file_name)
def _count_genbank_sequences(self, filename):
"""
This is not a perfect definition, but should suffice for general usage. It fails to detect any
errors that would result in parsing errors like incomplete files.
"""
# Specification for the genbank file format can be found in
# ftp://ftp.ncbi.nih.gov/genbank/gbrel.txt
# in section 3.4.4 LOCUS Format
count = 0
with open(filename) as gbk:
for line in gbk:
if line[0:5] == 'LOCUS':
count += 1
return count
Composite Datatypes¶
Composite datatypes can be used as a more structured way to contain individual history items which are composed of multiple files. The Rgenetics package for Galaxy has been implemented using Composite Datatypes; for real-life examples, examine the configuration files (particularly lib/galaxy/datatypes/genetics.py) included with the distribution.
In composite datatypes, there is one “primary” data file and any number of specified “component” files. The primary data file is a base dataset object and the component files are all located in a directory associated with that base dataset. There are two types of composite datatypes: basic and auto_primary_file. In basic composite datatypes, the primary data file must be written by tools or provided directly during upload. In auto_primary_file composite datatypes, the primary data file is generated automatically by the Framework; all tool-writable or uploadable files are stored in the directory associated with the primary file.
Creating Composite Datatypes¶
A datatype can be set to composite by setting the composite_type flag. There are 3 valid options:
- None (not a composite datatype)
basicauto_primary_file
Defining Basic Composite Datatypes¶
The example below defines a basic composite datatype which is composed of 2 component files along with a tool-written or user-uploaded primary file. In this example the primary file is a report summarizing the results. The two component files (results.txt, results.dat) contain the results; results.txt is a text file and is handled as such during upload whereas results.dat is flagged as binary, allowing a binary file to be uploaded for that component. During upload, 3 sets of file upload/paste interfaces are provided to the user. A file must be provided for the primary (index) file as well as results.txt; results.dat is flagged as optional so may be left blank.
class BasicComposite(Text):
composite_type = 'basic'
def __init__ (self, **kwd):
Text. __init__ (self, **kwd)
self.add_composite_file('results.txt')
self.add_composite_file('results.dat', is_binary = True, optional = True)
These files can be specified on the command line in the following fashion:
<command>someTool.sh '$input1' '${os.path.join(input1.extra_files_path, 'results.txt')}'
'${os.path.join(input1.extra_files_path, 'results.dat')}' '$output1'</command>
If a tool is aware of the file names for a datatype, then only input1.extra_files_path needs to be provided.
There are cases when it is desirable for the composite filenames to have varying names, but be of a similar form; for an example of this see Rgenetics below.
Defining Auto Primary File Composite Datatypes¶
The example below defines an auto primary file composite datatype which is composed of 2 component files along with a framework generated file. In this example the primary file is an html page containing download links to the individual result files. The two component files (results.txt, results.dat) contain the results; results.txt is a text file and is handled as such during upload whereas results.dat is flagged as binary, allowing a binary file to be uploaded for that component. During upload, 2 sets of file upload/paste interfaces are provided to the user. A file must be provided for results.txt whereas results.dat is flagged as optional so may be left blank.
In this composite type, the primary file is generated using the generate_primary_file method specified in the datatype definition. The generate_primary_file method here loops through the defined components and creates a link to each.
class AutoPrimaryComposite(Text):
composite_type = 'auto_primary_file'
def __init__ (self, **kwd):
Text. __init__ (self, **kwd)
self.add_composite_file('results.txt')
self.add_composite_file('results.dat', is_binary=True, optional=True)
def generate_primary_file(self, dataset=None):
rval = ['<html><head><title>Files for Composite Dataset (%s)</title></head><p/>This composite dataset is composed of the following files:<p/><ul>' % (self.file_ext)]
for composite_name, composite_file in self.get_composite_files(dataset=dataset).iteritems():
opt_text = ''
if composite_file.optional:
opt_text = ' (optional)'
rval.append('<li><a href="%s">%s</a>%s' % (composite_name, composite_name, opt_text))
rval.append('</ul></html>')
return "\n".join(rval)
These files can be specified on the command line in the following fashion:
<command>someTool.sh ${os.path.join(input1.extra_files_path, 'results.txt')} ${os.path.join(input1.extra_files_path, 'results.dat')} $output1</command>
Advanced Topics: Rgenetics Example¶
Rgenetics datatypes are defined as composite datatypes. The tools in this package rely heavily on the contents of a filename for analysis and reporting. In this case it is desirable for the filenames of the components to vary slightly, but maintain a common form. To do this, we use a special metadata parameter that can only be set at dataset creation (i.e. upload). This example uses the metadata parameter base_name to specify part of the components’ filenames.
class Lped(Text):
MetadataElement(name="base_name", desc="base name for all transformed versions of this genetic dataset", default="galaxy", readonly=True, set_in_upload=True)
composite_type = 'auto_primary_file'
file_ext="lped"
allow_datatype_change = False
def __init__(self, **kwd):
Rgenetics.__init__(self, **kwd)
self.add_composite_file('%s.ped', description='Pedigree File', substitute_name_with_metadata='base_name', is_binary=True)
self.add_composite_file('%s.map', description='Map File', substitute_name_with_metadata='base_name', is_binary=True)
def generate_primary_file(self, dataset=None):
rval = ['<html><head><title>Files for Composite Dataset (%s)</title></head><p/>This composite dataset is composed of the following files:<p/><ul>' % (self.file_ext)]
for composite_name, composite_file in self.get_composite_files(dataset=dataset).iteritems():
opt_text = ''
if composite_file.optional:
opt_text = ' (optional)'
rval.append('<li><a href="%s">%s</a>%s' % (composite_name, composite_name, opt_text))
rval.append('</ul></html>')
return "\n".join(rval)
The file specified as %s.ped is found at os.path.join(input1.extra_files_path, '%s.ped' % input1.metadata.base_name).
It should be noted that changing the datatype of datasets which use this substitution method will cause an error if the metadata parameter ‘base_name’ does not exist in a datatype that the dataset is set to. This is because the value within ‘base_name’ will be lost – if the datatype is set back to the original datatype, the default metadata value will be used and the filenames might not match the basename.
To prevent this from occurring, set allow_datatype_change to False. The dataset’s datatype will not be able to be changed.
Galaxy Tool Shed - Data Types¶
Note: These are deprecated and shouldn’t be used.
Including custom data types that subclass from Galaxy data types in the distribution¶
If your repository includes tools that require data types that are not defined in the Galaxy distribution, you can include the required data types in the repository along with your tools, or you can create a separate repository to contain them. The repository must include a file named datatypes_conf.xml, which is modeled after the file named datatypes_conf.xml.sample in the Galaxy distribution. This section describes support for including data types that subclass from data types in the Galaxy distribution. Refer to the next section for details about data types that use your own custom class modules included in your repository. An example of this is the datatypes_conf.xml file in the emboss_datatypes repository in the main Galaxy tool shed, shown below.
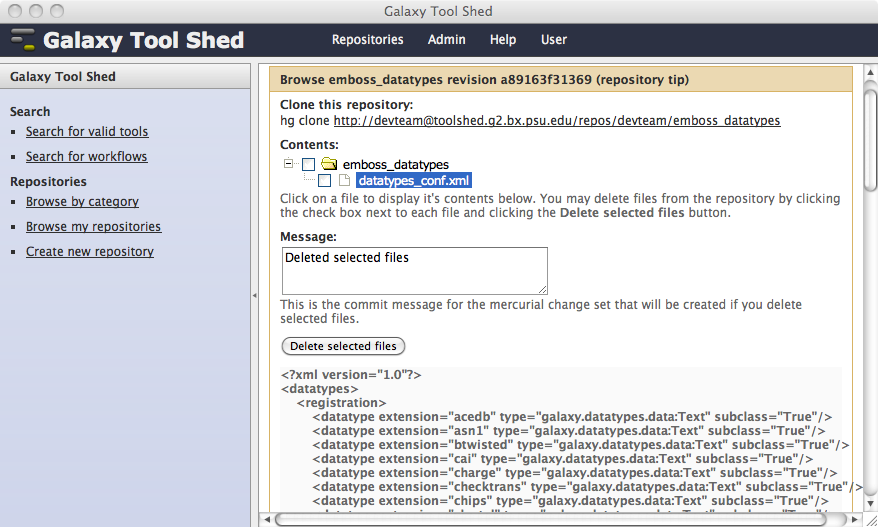
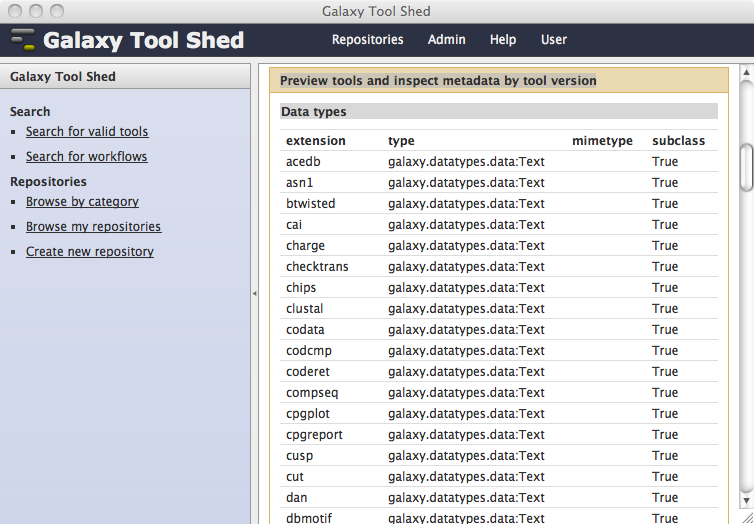
Tool shed repositories that include valid datatypes_conf.xml files will display the data types in the Preview tools and inspect metadata by tool version section of the view or manage repository page.
Including custom data types that use class modules contained in your repository¶
Including custom data types that use class modules included in your repository is a bit tricky. As part of your development process for tools that use data types that fall into this category, it is highly recommended that you host a local Galaxy tool shed. When your newly developed tools have proven to be functionally correct within your local Galaxy instance, you should upload them, along with all associated custom data types files and modules to your local tool shed to ensure that everything is handled properly within the tool shed. When your local tool shed repository is functionally correct, install your repository from your local tool shed to a local Galaxy instance to ensure that your tools and data types properly load both at the time of installation and when you stop and restart your Galaxy server. You should not upload your tools to the main Galaxy tool shed until you have confirmed that everything works by following these steps.
To illustrate how this works, we’ll use the gmap repository in the main Galaxy tool shed as an example. The datatypes_conf.xml file included in this repository looks something like the following. You’ll probably notice that this file is modeled after the datatypes_conf.xml.sample file in the Galaxy distribution, but with some slight differences.
Notice the <datatypes_files> tag set. This tag set contains <datatype_file> tags, each of which refers to the name of a class module file name within your repository (in this example, there is only one file named gmap.py), which contains the custom data type classes you’ve defined for your tools.
In addition, notice the value of each type attribute in the <datatype> tags. The : separates the class module included in the repository (in this example, the class module is gmap) from the class name (GmapDB, IntervalAnnotation, etc.). It is critical that you make sure your datatype tag definitions match the classes you’ve defined in your class modules or the data type will not properly load into a Galaxy instance when your repository is installed.
<?xml version="1.0"?>
<datatypes>
<datatype_files>
<datatype_file name="gmap.py"/>
</datatype_files>
<registration>
<datatype extension="gmapdb" type="galaxy.datatypes.gmap:GmapDB" display_in_upload="False"/>
<datatype extension="gmapsnpindex" type="galaxy.datatypes.gmap:GmapSnpIndex" display_in_upload="False"/>
<datatype extension="iit" type="galaxy.datatypes.gmap:IntervalIndexTree" display_in_upload="True"/>
<datatype extension="splicesites.iit" type="galaxy.datatypes.gmap:SpliceSitesIntervalIndexTree" display_in_upload="True"/>
<datatype extension="introns.iit" type="galaxy.datatypes.gmap:IntronsIntervalIndexTree" display_in_upload="True"/>
<datatype extension="snps.iit" type="galaxy.datatypes.gmap:SNPsIntervalIndexTree" display_in_upload="True"/>
<datatype extension="gmap_annotation" type="galaxy.datatypes.gmap:IntervalAnnotation" display_in_upload="False"/>
<datatype extension="gmap_splicesites" type="galaxy.datatypes.gmap:SpliceSiteAnnotation" display_in_upload="True"/>
<datatype extension="gmap_introns" type="galaxy.datatypes.gmap:IntronAnnotation" display_in_upload="True"/>
<datatype extension="gmap_snps" type="galaxy.datatypes.gmap:SNPAnnotation" display_in_upload="True"/>
</registration>
<sniffers>
<sniffer type="galaxy.datatypes.gmap:IntervalAnnotation"/>
<sniffer type="galaxy.datatypes.gmap:SpliceSiteAnnotation"/>
<sniffer type="galaxy.datatypes.gmap:IntronAnnotation"/>
<sniffer type="galaxy.datatypes.gmap:SNPAnnotation"/>
</sniffers>
</datatypes>
Modules that include custom datatype class definitions cannot use relative import references for imported modules. To function correctly when your repository is installed in a local Galaxy instance, your class module imports must be defined as absolute from the galaxy subdirectory inside the Galaxy root’s lib subdirectory. For example, assume the following import statements are included in our example gmap.py file. They certainly work within the Galaxy development environment when the gmap tools were being developed.
import data
from data import Text
from metadata import MetadataElement
However, the above relative imports will not work when the gmap.py class module is installed from the Tool Shed into a local Galaxy instance because the modules will not be found due to the use of the relative imports. The developer must use the following approach instead. Notice that the imports are written such that they are absolute relative to the ~/lib/galaxy subdirectory.
import galaxy.datatypes.data
from galaxy.datatypes.data import Text
from galaxy.datatypes.metadata import MetadataElement
The use of <converter> tags contained within <datatype> tags is supported in the same way they are supported within the datatypes_conf.xml.sample file in the Galaxy distribution.
<datatype extension="ref.taxonomy" type="galaxy.datatypes.metagenomics:RefTaxonomy" display_in_upload="true">
<converter file="ref_to_seq_taxonomy_converter.xml" target_datatype="seq.taxonomy"/>
</datatype>
Including datatype converters and display applications¶
To include your custom datatype converters or display applications, add the appropriate tag set to your repository’s datatypes_conf.xml file in the same way that they are defined in the datatypes_conf.xml.sample file in the Galaxy distribution.
If you include datatype converter files in your repository, all files (the disk file referred to by the value of the “file” attribute) must be located in the same directory in your repository hierarchy. Similarly, your datatype display application files must all be in the same directory in your repository hierarchy (although the directory can be a different directory from the one containing your converter files). This is critical because the Galaxy components that load these custom items assume each of them are located in the same directory.